 TARGETING ENERGETICS TO KILL TUBERCULOSIS
TARGETING ENERGETICS TO KILL TUBERCULOSIS
Tuberculosis caused more deaths in 2019 than any other infectious disease (~1.3 million) and latently infects a third of the world’s population, predominantly in the developing world. A major barrier to the disease’s treatment is the capacity of its causative agent to persist in host tissues. This contributes to the difficulty in clearing infections and the development of antibiotic resistance due to fractional killing. Improved treatment depends on understanding and targeting the mechanisms that facilitate persistence and resistance.
To achieve this, we study the metabolic basis of mycobacterial persistence. We investigate the physiological roles and biochemical mechanisms of multiple redox enzymes implicated in persistence. The end goal is to validate novel targets for pharmaceutical development.
Major projects
1. Carbon monoxide: poison or fuel for Mycobacterium tuberculosis? (NHMRC EL2 Fellowship, 2020 – 2024)
2. Development of a cofactor biosynthesis pathway as an antitubercular drug target (NHMRC New Investigator Grant, 2018 – 2021)
3. Prediction and alleviation of clinical nitroimidazole prodrug resistance (NHMRC Project Grant, 2018 – 2021)
4. Systematic testing of novel antimicrobial compounds and surfaces against pathogen panels (Centre to Impact AMR Project)
Research team
Staff: Dr Paul Cordero (postdoc), Ashleigh Kropp (research assistant)
Students: Katie Bayly (PhD), David Gillett (PhD), Anjali Lobo (Honours)
Collaborators: Prof Gregory Cook, Dr Rhys Grinter, Prof Ross Coppel, Prof Colin Jackson, A/Prof Nick West, Prof Ben Mirais, A/Prof Max Cryle, Dr Ghader Bashiri
Previous findings
Our work has revealed the unusual redox cofactor F420 is critical for redox homeostasis and drug metabolism in mycobacteria (JMB 2015, AEM 2016, Frontiers 2017, PLOS Pathogens 2020). We have resolved its biosynthesis pathway and solved crystal structures of its enzymes as a basis for drug development (ISME 2017a, Nature Comms 2019, mSystems 2020). Understanding this pathway is also critical to preempt resistance to clinical prodrugs pretomanid and delamanid, which are both activated by F420 (MMBR 2016, PLOS Pathogens 2020).
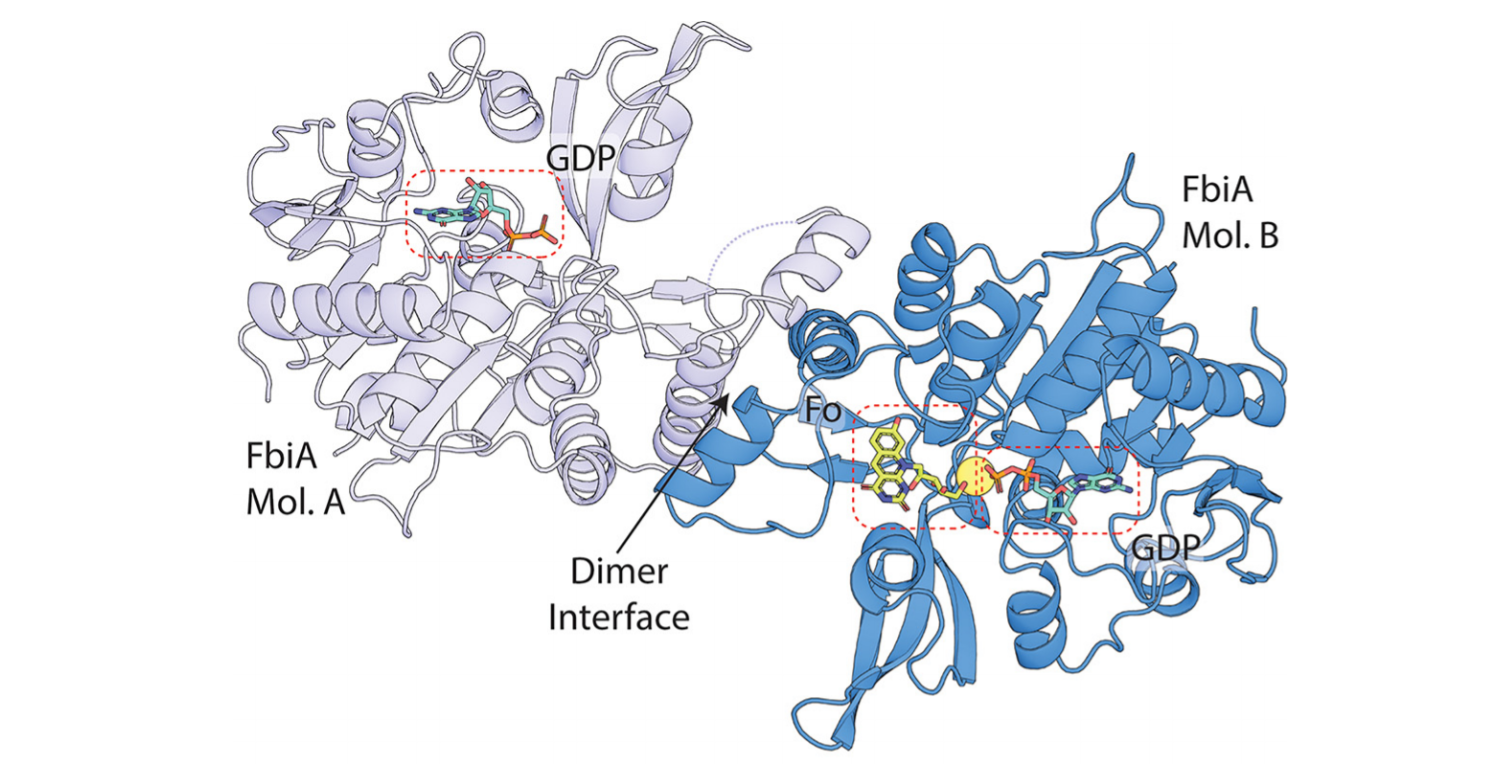
Figure: Example of our work on mycobacterial persistence. After resolving the enigmatic biosynthesis pathway of F420, we solved substrate- and product-bound structures of the key enzyme FbiA, revealing formation of the novel intermediate dehydro-F420-0 (mSystems 2020).
We have also shown that mycobacteria are more energetically flexible than previously thought. For example, mycobacteria adapt to energy starvation by consuming alternative energy sources such as carbon monoxide gas (ISME J 2019, bioRxiv 2021) and survive hypoxia by rewiring their redox metabolism (PNAS 2014, JBC 2019). This flexibility offers both opportunities and challenges for the development of metabolism as a target space for tuberculosis treatment (AMP 2014, Microbiol Spectrum 2017).